5.6 确定核心数据 EMP_identify_assay
传统的组学项目数据中经常存在着大量的稀疏特征(例如:微生物OTU/ASV注释表有着大量丰度极低的菌种),这些稀疏特征可能是由样本污染、建库错误、测序偏倚或者注释失败等原因导致。为了简化计算和减少这些稀疏特征的干扰,本模块提供了两种方法对原始数据进行过滤。
5.6.1 核心微生物数据
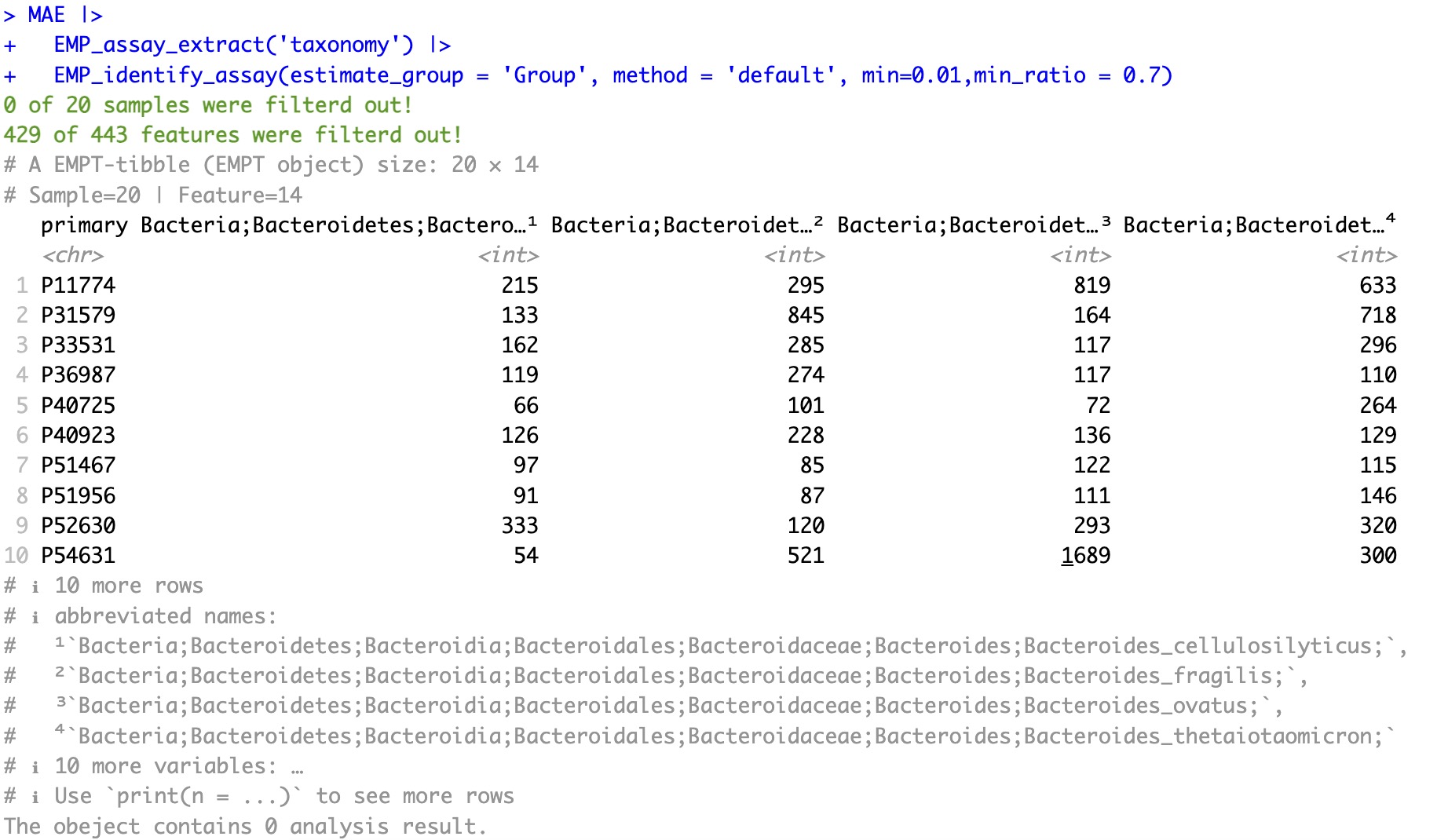
在微生物物种注释结果中,可以发现存在相当多的”稀有物种“(相对丰度较低或者在样本中出现的频率较低),对于识别组间具有差异性的物种造成极强的干扰,尤其是在筛选关键物种时,机器学习算法(例如:随机森林、LEFse)很容易将这些“稀有物种”识别为组间的“差异物种”。因此,有必要在正式分析前将这些“稀有物种”根据统一的标准进行过滤。EMP_identify_assay模块引入了两个重要参数帮助过滤:minnum(最小相对丰度)和min_ratio(最小占比)。
- 首先,在微生物数据中,低于指定“最小相对丰度”的任何丰度都会视为 0。(此举并不会真的修改原始数据为0)
- 随后,当某个物种在任意一组样本中高于”最小相对丰度“的样本数量占该分组下样本总数量的比例大于”最小占比“时,该物种就被认为是”核心物种“,而其余物种都被归类为“稀有物种”并被过滤掉。
🏷️示例:
使用模块EMP_assay_extract提取组学项目taxonomy的assay。使用模块EMP_identify_assay过滤获取“核心物种”,参数estimate_group指定将Group作为分组信息,参数min指定“最小相对丰度”为0.01,参数min_ratio指定最小占比为0.7。
注意:
①当输入的微生物物种注释数据为绝对丰度时,在计算过程中,模块
②这个阈值可以根据自己实验设计调整,没有固定的值。对于肠道微生物推荐使用最小相对丰度0.001和最小占比0.7。
①当输入的微生物物种注释数据为绝对丰度时,在计算过程中,模块
EMP_identify_assay会自动将其转换为相对丰度,以便在计算过程中进行过滤和获取核心物种,并根据过滤条件重新输出对应的绝对丰度的数据。②这个阈值可以根据自己实验设计调整,没有固定的值。对于肠道微生物推荐使用最小相对丰度0.001和最小占比0.7。
MAE |>
EMP_assay_extract('taxonomy') |>
EMP_identify_assay(estimate_group = 'Group', method = 'default', min=0.001,min_ratio = 0.7)
5.6.2 核心基因组数据
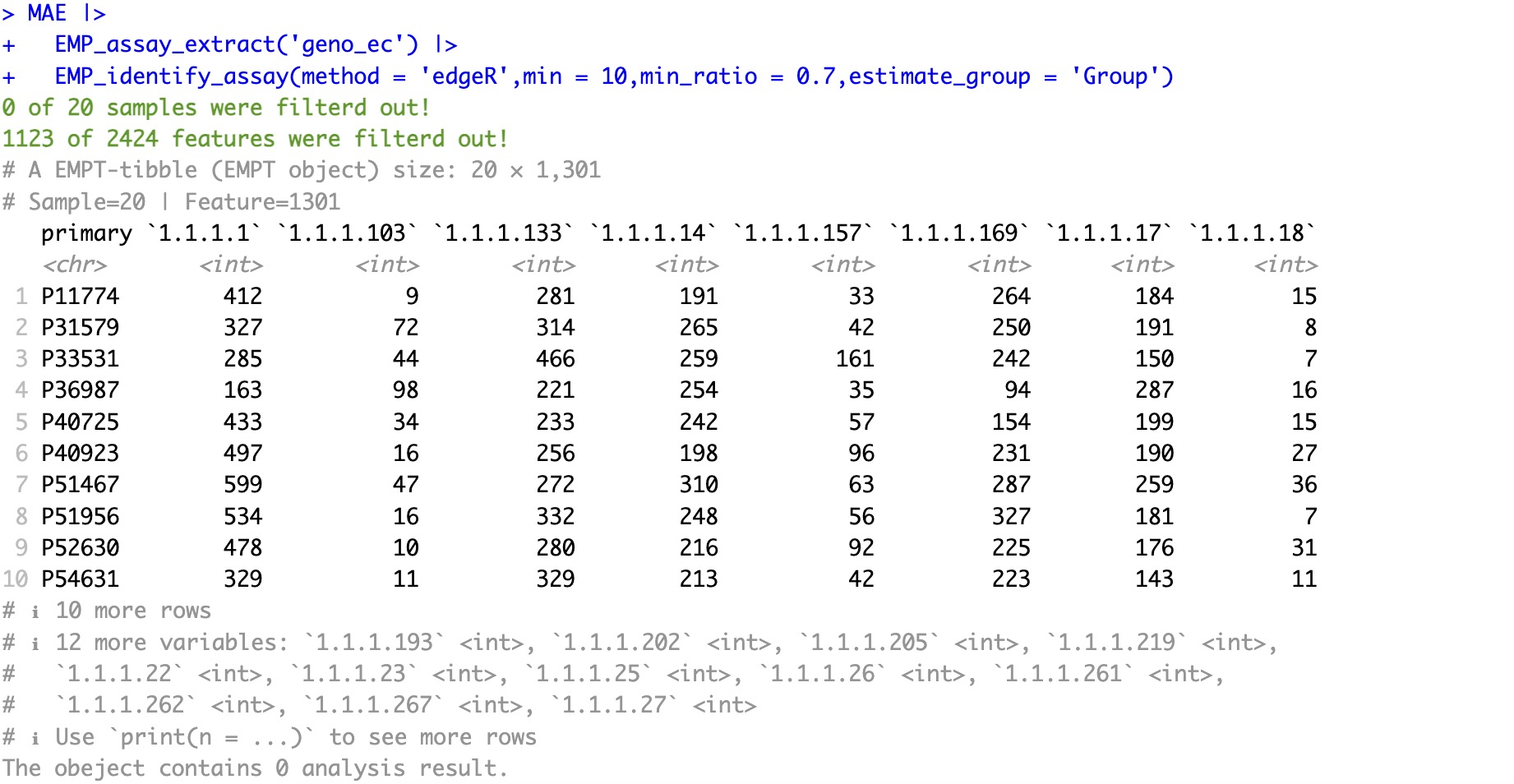
在基因组数据中,edgeR包提供了一种基于“最小丰度值”的过滤方法,本模块可以十分便捷地调用该方法对基因组/转录组等数据进行过滤。
🏷️示例:
MAE |>
EMP_assay_extract('geno_ec') |>
EMP_identify_assay(method = 'edgeR',min = 10,min_ratio = 0.7,estimate_group = 'Group')